Tutorials
Collection of step-by-descriptions that describe the most popular tools
Maybe you have a specific task in mind.
BLAST searches
Our Blast DBs are ordered into categories, namely by species common names. Further subcategories are introduced based on projects or special cultivars (e.g. „NCBI BLAST Rye Lo7“). Please choose your desired category. An overview of the blast DBs can be found here.
Example: Blastn against „Rye B chromosome“ transcript assembly
Open the category „NCBI BLAST Rye“ by left click. Now you can choose the blast tool, in this case „blastn“. The tool is displayed in the center panel.
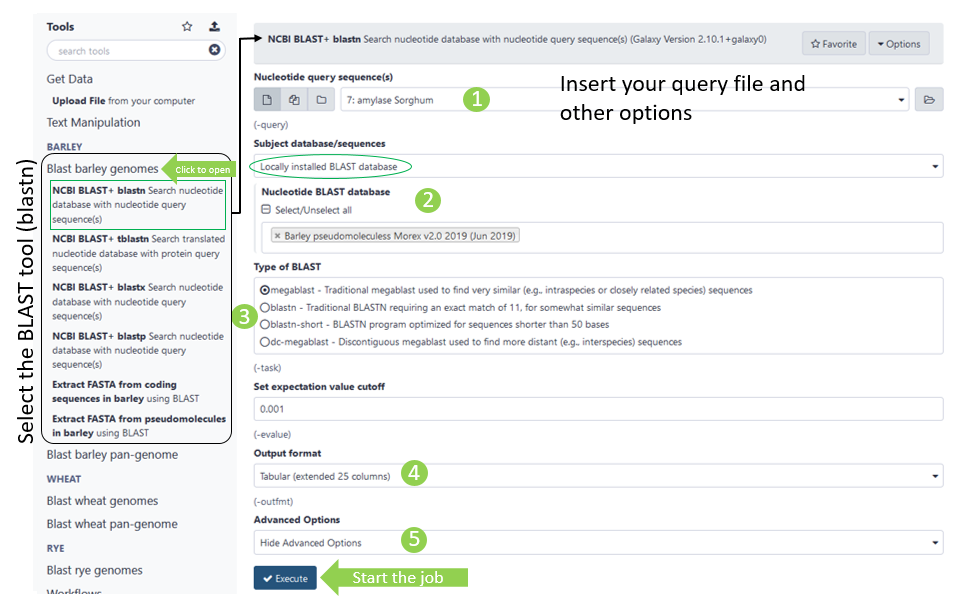
Once you opened to tool menu in the centre panel, you can make inputs and adjust other parametres to your need (the numbers refer to the green circles in figure above):
- All available input files (if in correct fasta format)
- Select DB to blast against
- The blast search algorithm (covered up)
- Opens additional parameters
- Output format
Output
The following Output formats are supported:
Default is the 25 column tabular output. Tabular output can be customized. The standard BLAST output is the alignment in pairs of query sequence and database match. For nucleotide, the matches are marked by a pipe symbol (“|”) in between query and database sequence. For protein, the identical matches are marked by letter code with “homologous” substitutions (determined by the scoring matrix used) marked by “+” symbol in a line between the query and the database sequence.
- Pairwise: Database alignments are shown in relation to the query sequence in pairwise fashion.
- Query-anchored: Database alignments are anchored and shown in relation to the query sequence. Identities are displayed as dots (.), with mismatches displayed as single letter abbreviations.
- Flat query-anchored: The 'flat' display shows inserts as deletions on the query. Identities are displayed as dots (.), with mismatches displayed as single letter abbreviations.
- XML Blast output: Output in XML format.
- Tabular: Simple tabular output with different alignment information separated according to tab delimited fields with field headers displayed at the top.
Options
BLAST+ offers wide range of options. You can find a detailed overview here.
Extract sequences based on BLAST hits
To retrieve sequences from your blast results two tools are available: based on the BLAST DBs you can choose between pseudomolecule assemblies or shorter target sequences like transcripts or EST collections The tools can be found in the specific NCBI Blast category for each species.
Example for NCBI BLAST+ Rye:
-“Blast pseudomolecules in Rye” to get fasta files of range in subject
-“Blast transcripts, ESTs in Rye to get fasta files of complete subject
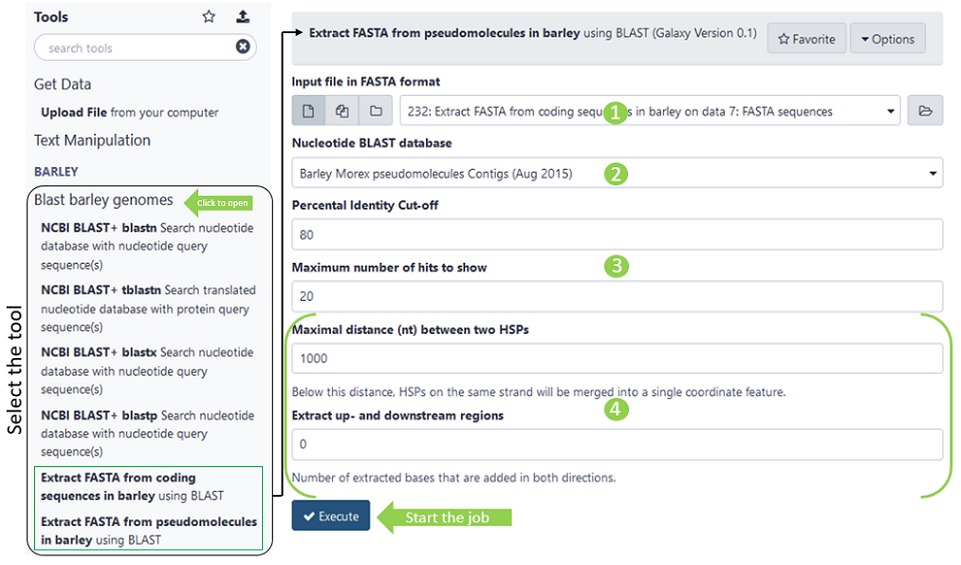
Once you opened to tool menu in the centre panel, you can make inputs and adjust other parametres to your need (the numbers refer to the green circles in figure above):
- All available input files (if in correct fasta format)
- Select DB to blast against
- Choose percental identity threshold and maximum number of hits parameter passed to blast algorythm
- Additional parameters for very long sequences, see description below*
*Options only available for Blast pseudomolecules in Rye!
Input
Just like starting a normal blast tool, the input should be a fasta file with your query sequences. You could also use a multiple fasta file. In this case, be aware of the output HSP limit. You may not receive all possible valid hits. To overcome this limitation, split you multiple fasta into single files and choose batch input.
Output
As an output you will receive two files: A 13-columns tabular Blast file and most probably a fasta file with sequences. Files remain empty, if the blast search returns no hits at all.
The tool “Blast transcripts, ESTs in [species]” will return the complete sequence of the blast subject. If you blast for pseudomolecule sequences, the coordinates of the hit will be used to extract the sequence. To modify the fasta sequence, you can use the following options.
Options
The following options are only valid for the tool “Blast pseudomolecules in [species]”:
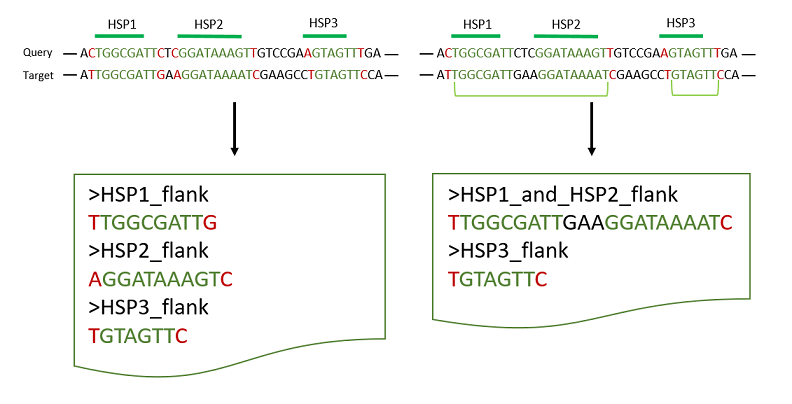
Merge HSPs
When blasting a gene to genomic sequence, the resulting alignment might be gapped in some cases, e.g. introns or general sequence deviations.
The resulting output consists of so called HSPs (High-scoring Segment Pair). To combine multiple neighbored HSPs and finally extract the maximal range of the putative hit sequence, bedtools merge parameter -d is used. It defines the maximum distance within a join will be carried out. By default, this value is ‚0‘, meaning that only overlapping HSPs will be merged into one feature.

The resulting fasta file depends on the parameter
- –d 0 will output each HSP seperately (on the left).
- –d 5 will join all single HSPs, that share a maximum distance of 5 nucleotides. In here HSP1 and two will be merged, whereas HSP3 remains as individual feature because of the >5 nt spacing (right side).
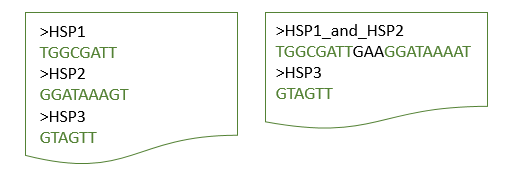
Extract flanking sequences
If you wish to extract the flanking sequences of targets additionally, you can use the –l parameter. It defaults to 0, meaning the exact start and stop coordinates of your hit will be cut.
The number of flanking nucleotides will be the same for up- and downstream region.
If the parameter –l 1 is added to previous example, one flanking base will be attched in each direction. Depending on the parameter ‘–d’, HSPs might be merged before or treated separately (see figure below).